Data Organization and Management in Nanopore Sequencing
The importance of good data management
Effective data management is essential for ensuring the integrity, reproducibility, and long-term accessibility of sequencing data. Following best practices for organizing, storing, and documenting sequencing data benefits both research efficiency and educational experiences. In genomics, the volume of data generated can be substantial, making structured data organization critical.
A guiding framework for good data stewardship is the FAIR Principles:
- Findable – Data should be easily discoverable using standardized metadata.
- Accessible – Data should be retrievable via established repositories and protocols.
- Interoperable – Data should be compatible with various analysis tools and software.
- Reusable – Data should be well-documented and structured to allow for future use.
For educators, introducing students to data hygiene—including proper file naming, directory structuring, and metadata collection—helps instill best practices that are critical for careers in bioinformatics, computational biology, and genomics.
Instruction tip
Managing Student-Generated Data
Managing student-generated data in a Nanopore sequencing course requires careful planning to ensure smooth data sharing, organization, and long-term usability. Educators should establish a clear data-sharing strategy that facilitates seamless file exchange between instructors and students, particularly given the large file sizes and/or large number of files associated with sequencing data. Cloud storage solutions like CyVerse, Google Drive, or institutional servers may be useful for storing and distributing datasets, but file size limitations and access controls should be considered. Additionally, educators should anticipate the scientific value of student-generated data—insisting on good data practices from the beginning can ensure that sequencing results are valid, reproducible, and valuable in aggregate.
Developing a classroom data management plan can help structure how data are named, stored, and tracked across multiple course sections, lab groups, or project days. Establishing standardized file-naming conventions that incorporate identifiers such as course section, date, group number, and experiment type will allow educators to efficiently organize and retrieve data. Collecting consistent metadata across student projects is also essential if data are to be meaningfully analyzed or aggregated for further research or public sharing. Resources such as the FAIR Principles provide guidance on making data findable, accessible, interoperable, and reusable.
File and object names and metadata to keep track of from the start
- Project name: At the most encompassing level of organization, have a simple project name
- Students involved: Have a way of tracking students by more than give names and initials. Perhaps all students can be assigned a short unique identifier. You could consider having students create and use ORCIDs.
- Sample names: Have a way to name specific samples. The sample name might also include or be linked to acquisition information such as the date collected, or the location of sampling. If a single sampled is sampled multiple times, have a way to track and indicate this.
- Replicates: Have a way to indicate replication.
- Data directory structures: Consider how directories on storage platforms will be organized. Have rules for how folders should be named, when folders should be created, and what should be in them.
- Collision avoidance: Some files and folders will not have unique names. For example, every time you run a 16S workflow on MinKNOW, you will get folders named
barcode01,barcode02,barcode03... Know when you will encounter these and have a way to manage their organization, or if needed, renaming, to avoid confusion. - Software management: Have a way to keep track of the software versions of any software you will use.
While this list is not exhaustive, considering these possibilities ahead of time will make for smooth organization later.
By integrating good data hygiene and documentation practices, educators can help students engage with sequencing workflows in a way that mirrors professional research environments.
Data management plans
A Data Management Plan (DMP) is an essential document that outlines how data will be:
- Collected
- Stored and backed up
- Processed and analyzed
- Shared or archived
Many funding agencies require DMPs to ensure that research data remains well-documented and reusable. While formal DMPs may not be necessary for classroom projects, educators should emphasize the importance of:
- Keeping consistent directory structures
- Using clear, standardized file naming conventions
- Documenting key metadata
- Implementing proper backup procedures
Additional Reading
-
National Human Genome Research Institute. (n.d.). Genomic Data Science. National Human Genome Research Institute (NHGRI).
-
Oza, V. H., Whitlock, J. H., Wilk, E. J., Uno-Antonison, A., Wilk, B., Gajapathy, M., Howton, T. C., Trull, A., Ianov, L., Worthey, E. A., & Lasseigne, B. N. (2023). Ten simple rules for using public biological data for your research. PLoS Computational Biology, 19(1), e1010749. https://doi.org/10.1371/journal.pcbi.1010749.
-
Form, D., & Lewitter, F. (2011). Ten Simple Rules for Teaching Bioinformatics at the High School Level. PLoS Computational Biology, 7(10), e1002243. https://doi.org/10.1371/journal.pcbi.1002243.
-
Noble, W. S. (2009). A Quick Guide to Organizing Computational Biology Projects. PLoS Computational Biology, 5(7), e1000424. https://doi.org/10.1371/journal.pcbi.1000424.
Common file types in Nanopore sequencing
Nanopore sequencing generates multiple types of data files at different stages of the workflow. Below is an overview of the key file formats:
Raw data
- POD5 (.pod5)
[Documentation]
Current standard format for storing raw signal data from Nanopore sequencing. Replaces the older FAST5 format for improved performance and scalability. - FAST5 (.fast5) [Legacy]
Used in older Nanopore sequencing workflows to store raw signal data. This format is no longer in use, having been replaced by POD5.
Processed data
- FASTQ (.fastq)
[Documentation]
Primary file format for storing basecalled sequence reads, including nucleotide sequences and associated quality scores. - BAM (.bam) / CRAM (.cram)
[Documentation]
Binary formats used to store sequence alignments to a reference genome. CRAM is a more compressed version of BAM.
Reference data
- FASTA (.fasta / .fa)
[Documentation]
Stores nucleotide or protein sequences without quality scores. Often used for reference genomes or consensus sequences. - GFF/GTF (.gff / .gtf)
[Documentation]
Contains genome annotations, such as gene locations and functional elements.
Analysis and metadata
- CSV/TSV (.csv / .tsv)
Tabular files commonly used for summarizing sequencing statistics, barcode classifications, or other metadata. - JSON (.json)
Structured format for storing run configurations, analysis parameters, or experiment details. - VCF (.vcf)
Variant Call Format, used for storing genetic variations detected in sequencing data.
Reports
- HTML (.html)
Many analysis pipelines generate interactive reports in HTML format, offering visual summaries, charts, and metrics. This format is useful for sharing final results with collaborators or for archiving purposes.
Managing genomics data for long-term use
Metadata collection
Metadata provides essential context for sequencing datasets and is often required for submitting data to public repositories like the NCBI Sequence Read Archive (SRA). Key metadata elements may include:
- Sample ID (Unique identifier for the sample)
- Collection date (When the sample was obtained)
- Location (Geographic origin of the sample)
- Extraction and library prep methods (Protocols used)
- Sequencing platform and flow cell type
- Bioinformatics processing steps (Pipeline details, tools used)
Storage and backup strategies
- Primary Storage: A local high-capacity SSD (recommended 1TB or more) for sequencing and short-term analysis.
- Backup Storage: Redundant copies using external SSDs or network drives.
- Cloud Solutions: Platforms like Google Drive, CyVerse, or JetStream2 for scalable storage.
- Public Repositories: Depositing finalized datasets in SRA, EMBL-EBI ENA, or MG-RAST for community access.
Preparing data for public submission
- Ensure FASTQ files are properly formatted.
- Validate metadata against repository requirements.
- Use consistent file naming conventions (e.g.,
sampleID_experimentType_date.fastq). - Include a README file describing dataset contents and processing steps.
Quickstart: How to find and download Nanopore reads from the Sequence Read Archive (SRA)
The NCBI Sequence Read Archive (SRA) is a public repository that stores sequencing data, including Oxford Nanopore sequencing reads. This tutorial provides a step-by-step guide to searching for data, in this case, Actinomyces sequences generated using Oxford Nanopore technology and downloading a FASTQ file for further analysis. You can adapt the tutorial to search for data from your organism of interest and use those data for reanalysis and to practice your bioinformatics skills.
-
Open your web browser and visit the NCBI SRA website.
-
Click on the Advanced link beneath the search bar to access the detailed search options.
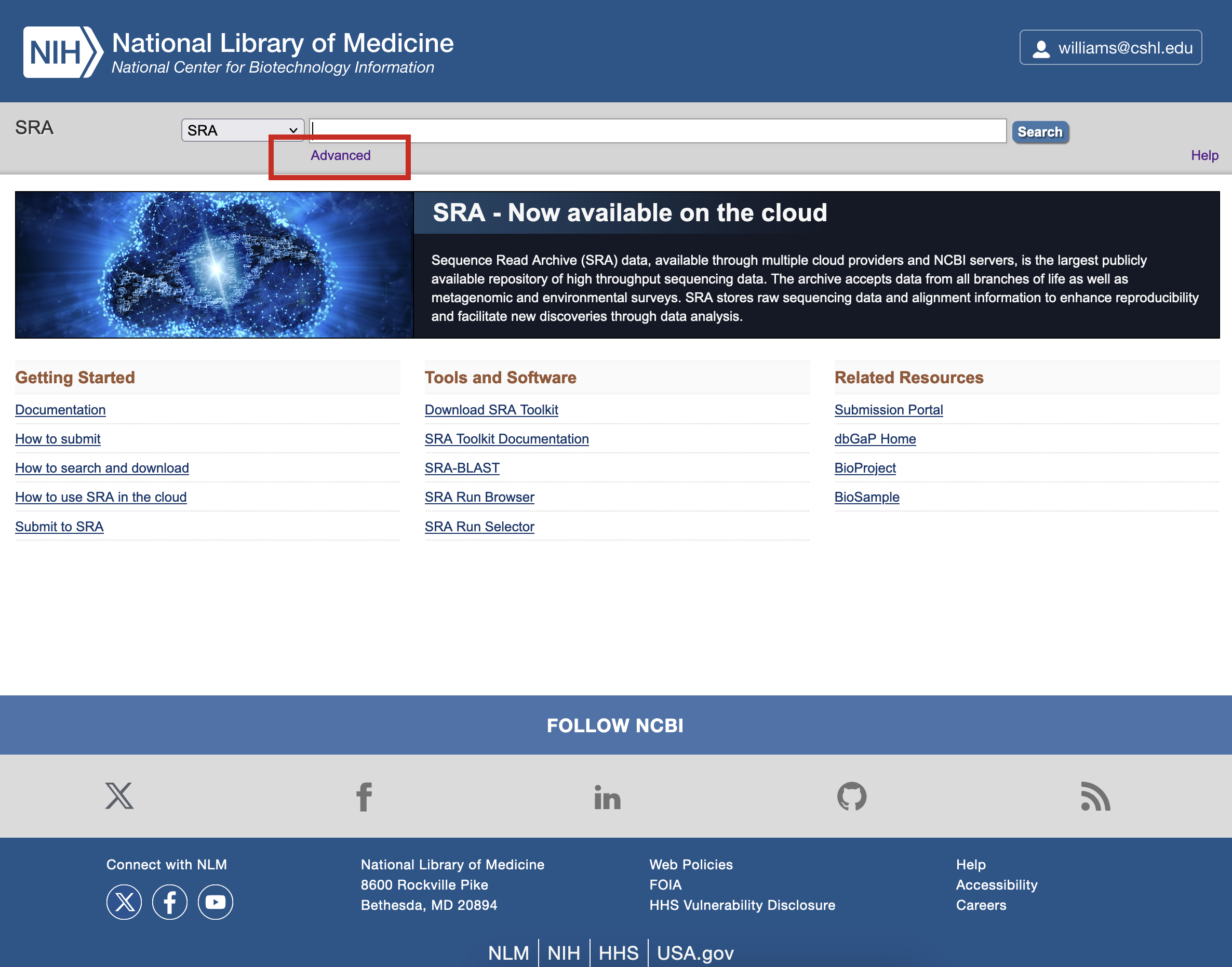
-
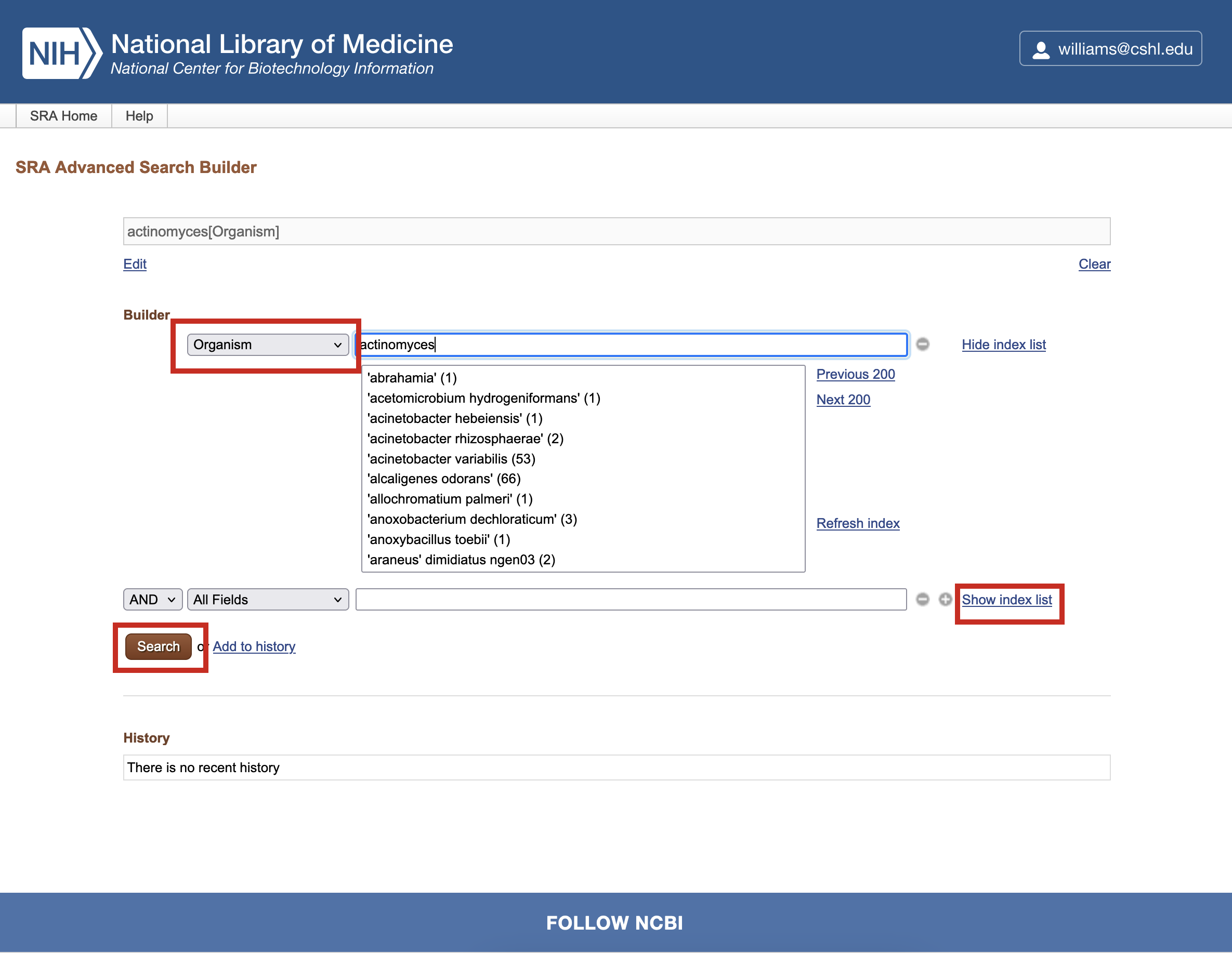
In the first box of the search Builder, type Actinomyces and click the Show index list link. The number in parentheses shows how many records match the term. You can click on the term of choice, in this case actinomyces (936), and select Organism from the dropdown to ensure that is the level being searched.
-
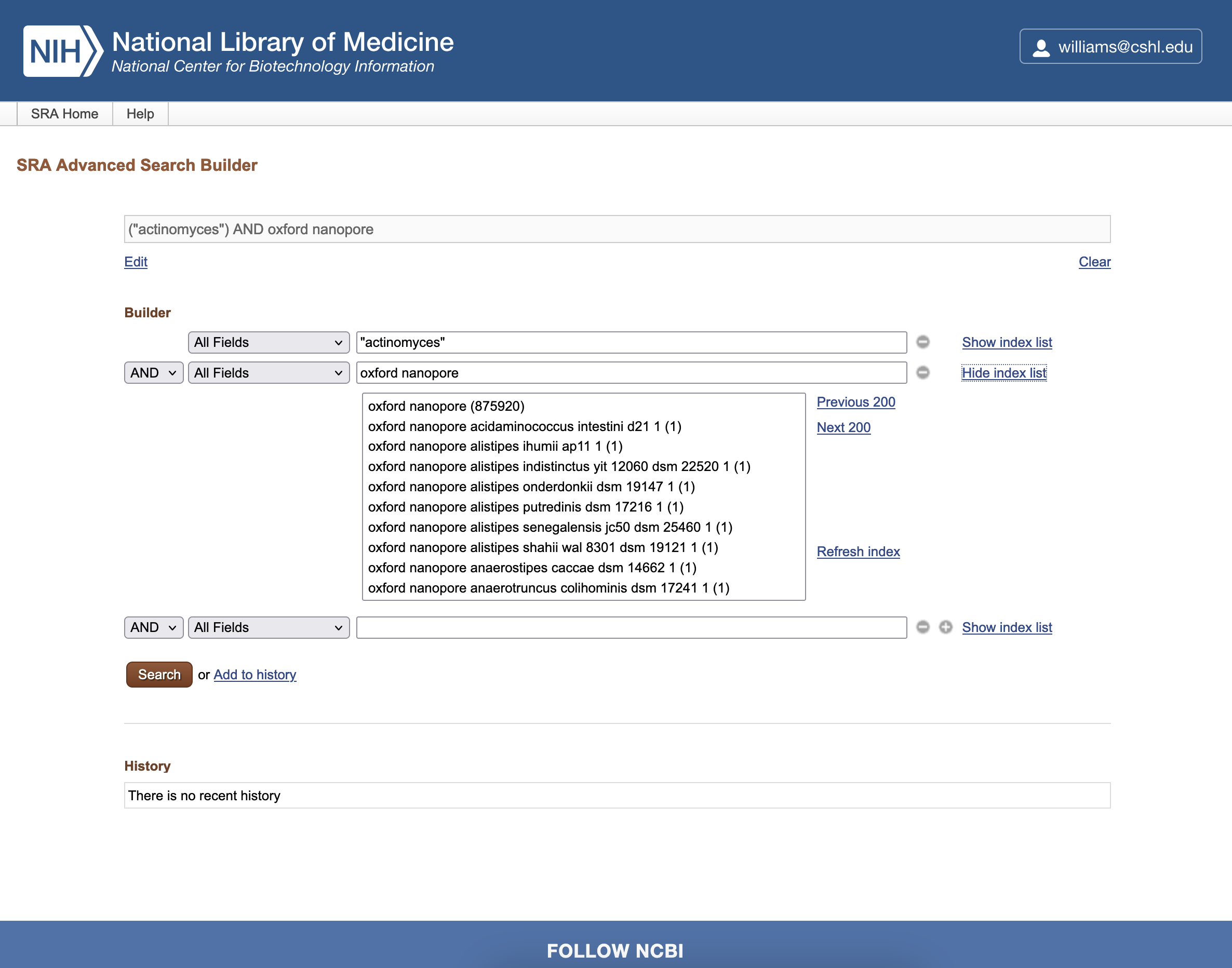
In the second box of the search Builder, type Oxford Nanopore and click the Show index list link. The number in parentheses shows how many records match the term. You can click on the term of choice, in this case oxford nanopore (875940), and select Platform from the dropdown to ensure that is the level being searched.
-
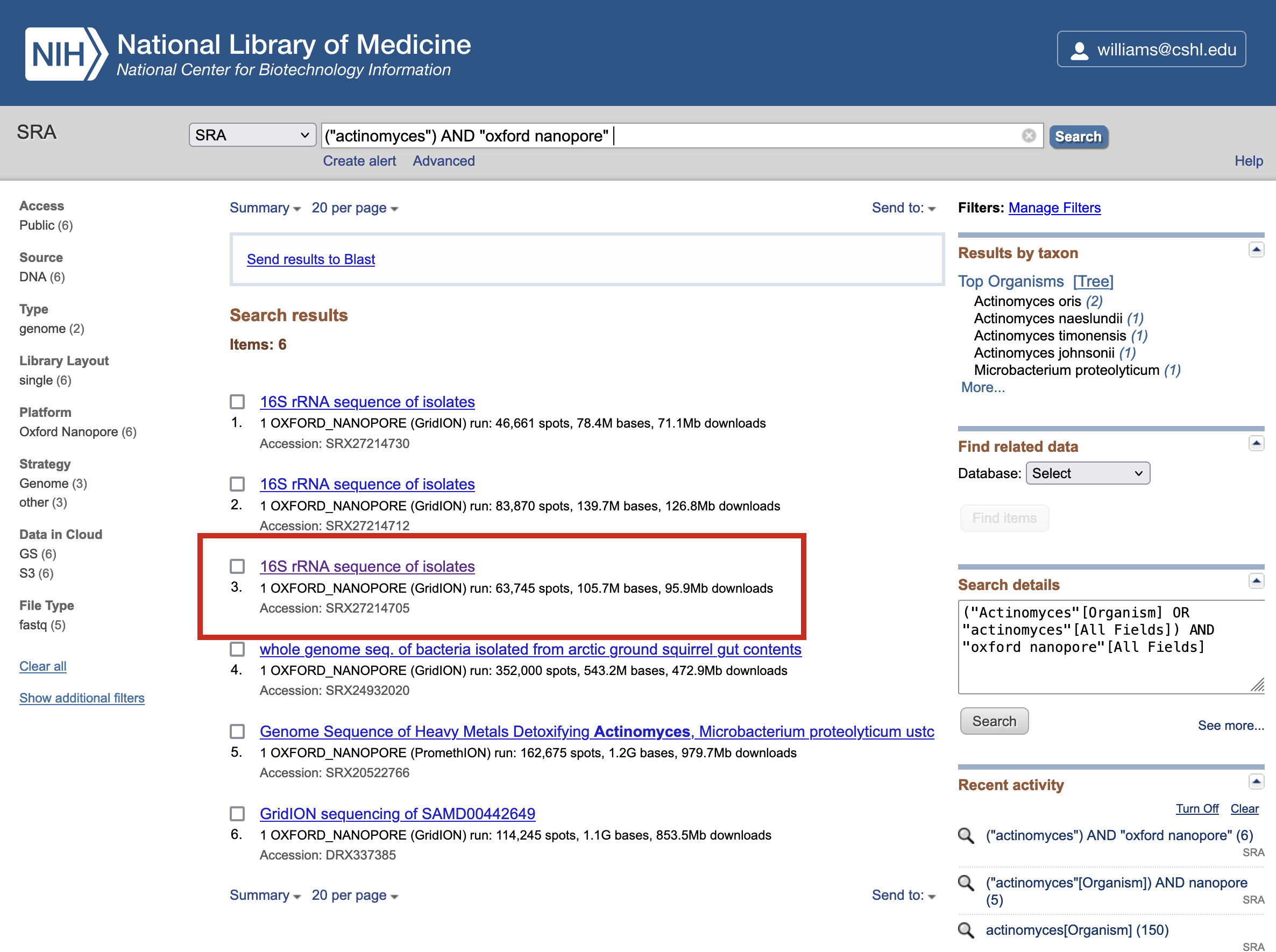
Click the Search button to retrieve relevant sequencing datasets.
-
Browse through the search results and look for a 16S sequencing dataset. In this case, we choose a selection marked 16S rRNA sequence of isolates. Note the number of bases (M - megabases, G - gigabases) and size of the dataset in Mb/Gb is shown, alerting you to the size of the potential download.; Click on a result to view the dataset details.
-
The results page will have various metadata including an abstract for the entry which may contain useful background on the experiment that produced the data.
Tip
The All experiments or All runs link will help you find connected datasets which may have come from other libraries or samples in this same experiment.
-
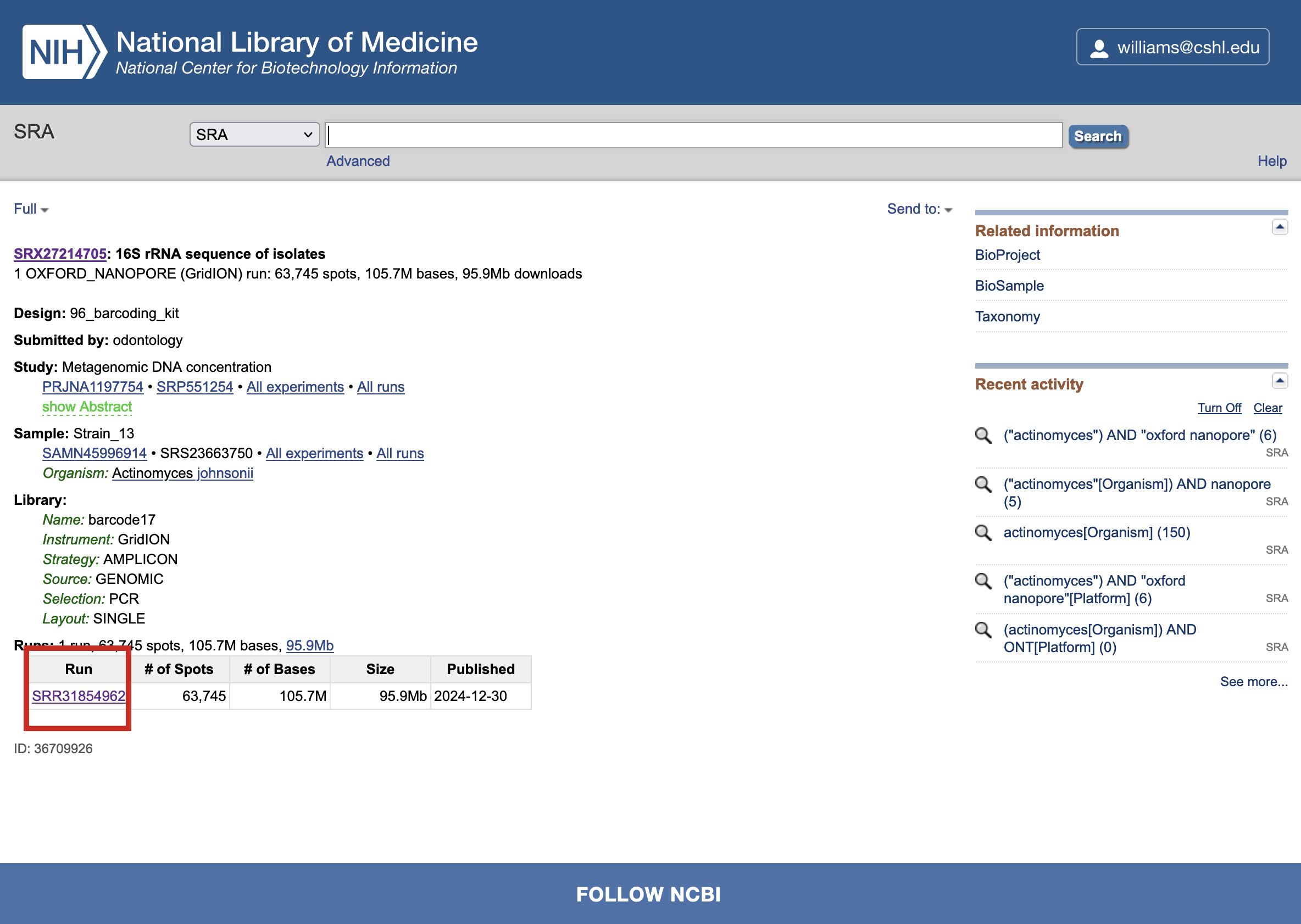
Locate the Runs table, which lists individual sequencing runs associated with the dataset.
-
Click on the Run Accession link (e.g., SRRXXXXXXX) of a single sequencing run to open its details page.
-
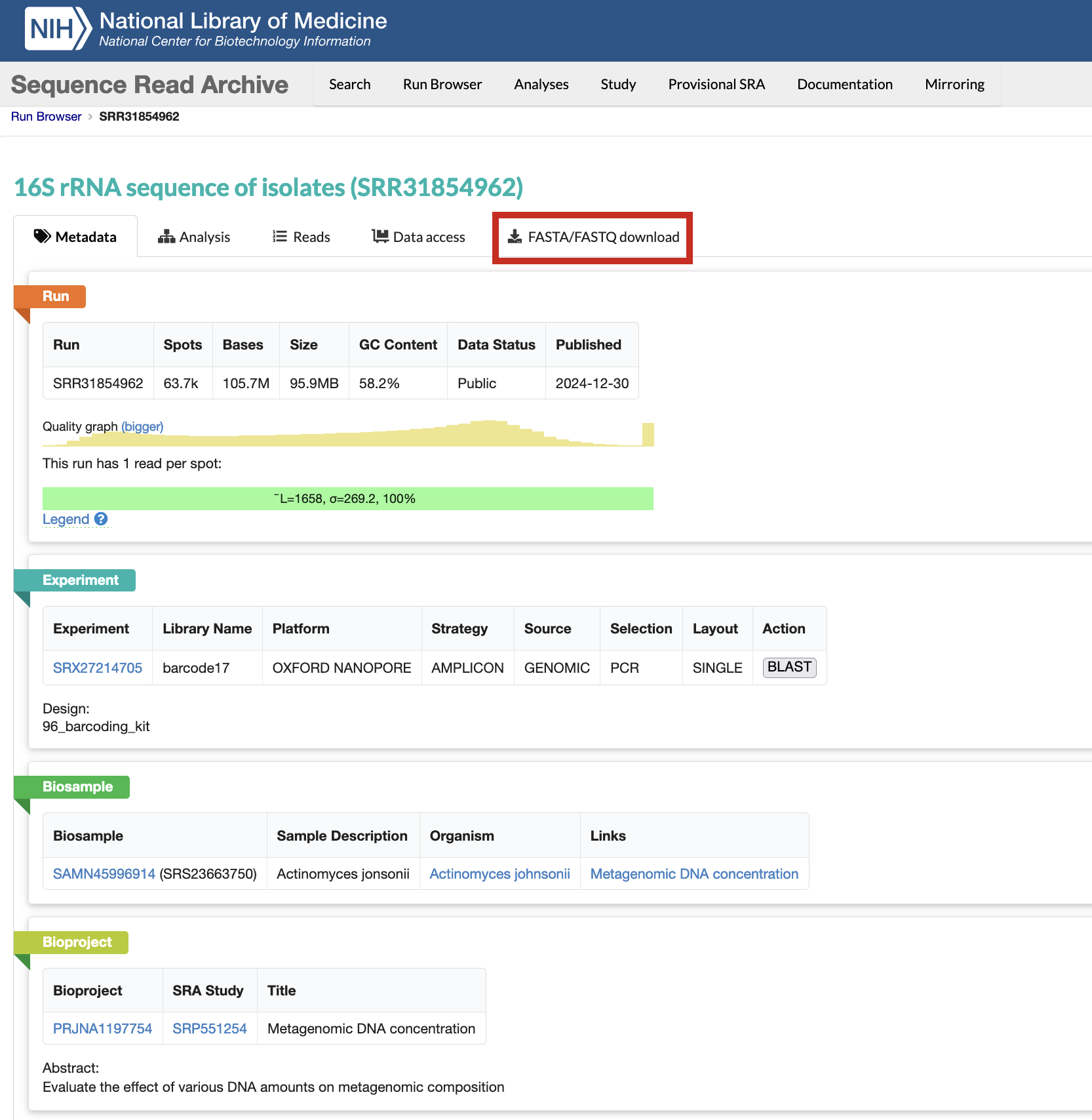
On the run browser page, click the FASTA/FASTQ download link.
-
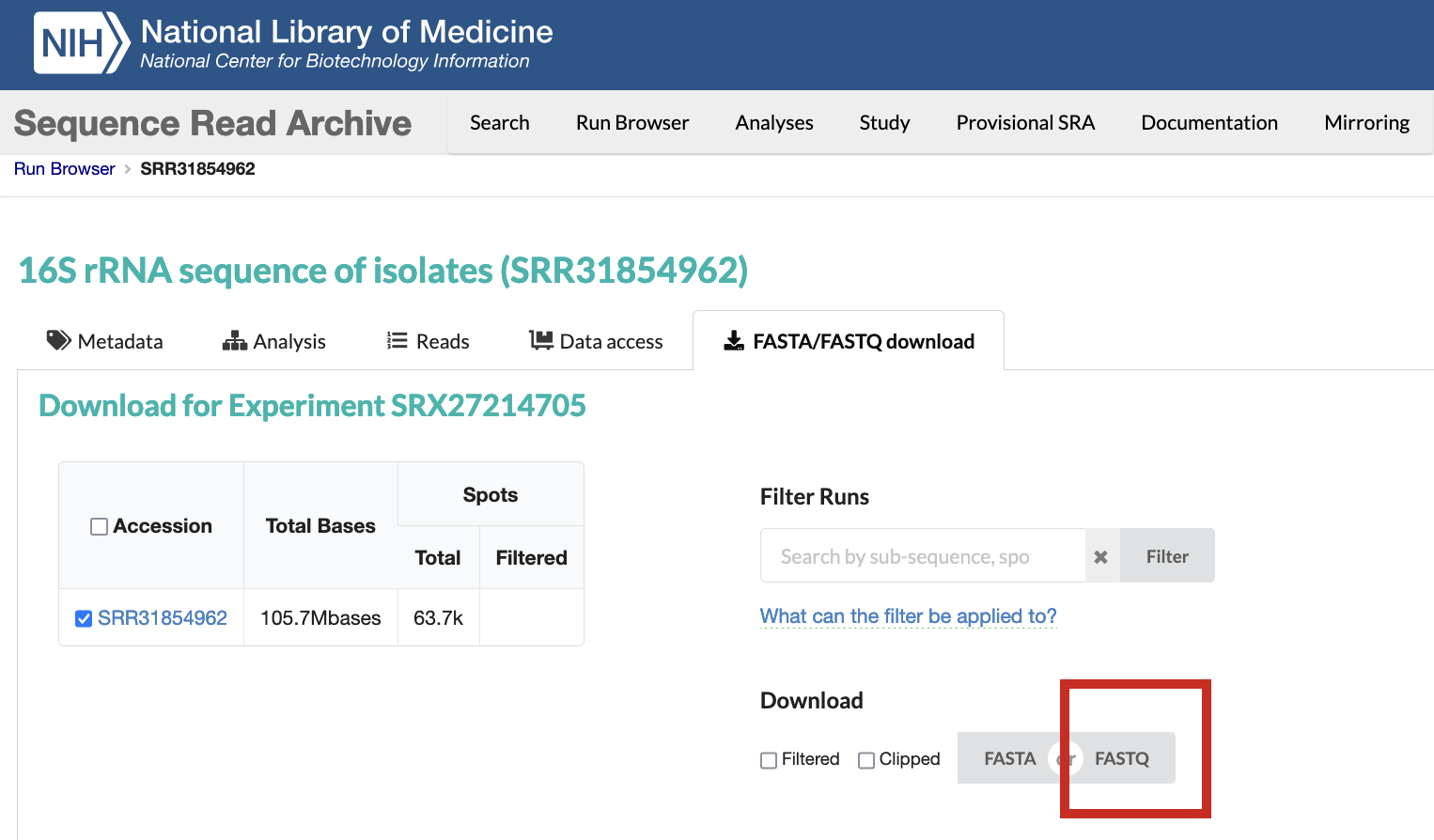
Under Download click on FASTQ to start a download for your file. The file will be in a compressed .fastq.gz format. This is a typical input for many downstream analyses.
Additional Notes
- The downloaded FASTQ file contains raw sequence reads and quality scores that can be used for taxonomic classification, assembly, or other bioinformatics workflows.
- Advanced users may explore SRA Toolkit, a command-line software package that enables more efficient searching, downloading, and processing of SRA data. The toolkit provides options such as
prefetchfor downloading andfastq-dumpfor extracting sequence reads. More details can be found in the SRA Toolkit documentation.
Comments and discussion
See recent comments or start a discussion on our Slack channel.